NDNA on Protein Interaction Networks
A 290-parameter genome learns which protein interactions matter for cancer, and the same genome works across breast, lung, colon, and prostate cancer.
The One-Paragraph Version
In Paper 1, NDNA was a tiny genome that grew the wiring of a neural network. In Paper 2, the same idea wired GPT-2. In Paper 3, we point the genome at a completely different kind of network: the human protein interaction network. Proteins inside a cell talk to each other; we have a map of who talks to whom. The genome learns which of those conversations matter for cancer. It rediscovers the Wnt signaling pathway and the cell cycle pathway entirely on its own (these are the two most studied drivers of cancer in biology). It also flags two genes that encode the most clinically used tumor markers in the world (the proteins doctors actually measure to detect cancer). And the same genome trained on breast cancer also works on lung, colon, and prostate cancer, with much more reliable results than random selection. A 290-parameter program found something general about how cancer works.
First, What Is a Protein Interaction Network?
Quick biology detour. You can skip this if you already know.
Your body is built out of proteins. Proteins are the workers inside every cell. They do almost everything: read your DNA, build new proteins, send signals, copy chromosomes, kill bad cells, repair damage, and so on. There are about 20,000 different proteins in a human cell, each with a specific job.
Proteins do not work alone. They physically touch each other. A protein binds to another protein, and that touching is how a signal gets passed, or a job gets done. Some proteins touch hundreds of partners; some touch only one or two.
If you draw all 20,000 proteins as dots and connect two dots whenever those proteins touch in real life, you get a giant tangled web. Scientists have spent decades mapping this web. The result is the protein interaction network, or PPI network.
Cancer is a disease of this network. When proteins start touching the wrong partners, or stop touching the right ones, cells start growing when they should not. Tumors form. Cancer kills.
The big question in computational cancer biology: of the millions of possible protein-protein interactions, which ones actually matter for cancer? Most do not. Identifying the ones that do is how we find new drug targets.
What We Did
We took a real protein interaction network from a public database called STRING, focused on the 5,000 most important genes for breast cancer, and ended up with a graph of about 17,700 connections.
Then we did the same thing NDNA always does: gave a 290-parameter genome the job of figuring out which connections matter.
This time the genome is not picking which neurons in an artificial network connect. It is picking which protein pairs, in a real biological network, are the ones that distinguish cancerous tissue from healthy tissue.
We trained it on breast cancer data from a public database called TCGA: gene activity levels measured in 1,200 patients (1,097 with tumors, 114 with healthy tissue). The genome looks at the network and decides: keep this protein-protein interaction, drop that one. The goal is to keep enough interactions that you can still tell tumor from healthy tissue, but no more.
The genome ended up keeping about 25% of the original interactions, the ones it judged most useful for the cancer-detection task.
What the Genome Found
This is the part that matters.
After training, we asked: of the interactions the genome decided to keep, how many overlap with biological pathways that cancer biologists already know are important?
A handful of biological pathways are central to most cancers. The two most studied:
- Wnt signaling is one of the master regulators of cell growth. When it gets stuck on, cells multiply uncontrollably. Wnt is a major driver of breast, colon, pancreatic, and many other cancers. Drug companies have entire programs targeting it.
- Cell cycle is the machinery that controls when a cell decides to divide. When this machinery breaks, you get cancer. Every cancer textbook starts here.
Both of these pathways are well documented and well understood. Did the genome rediscover them?
Yes. With strong statistical evidence.
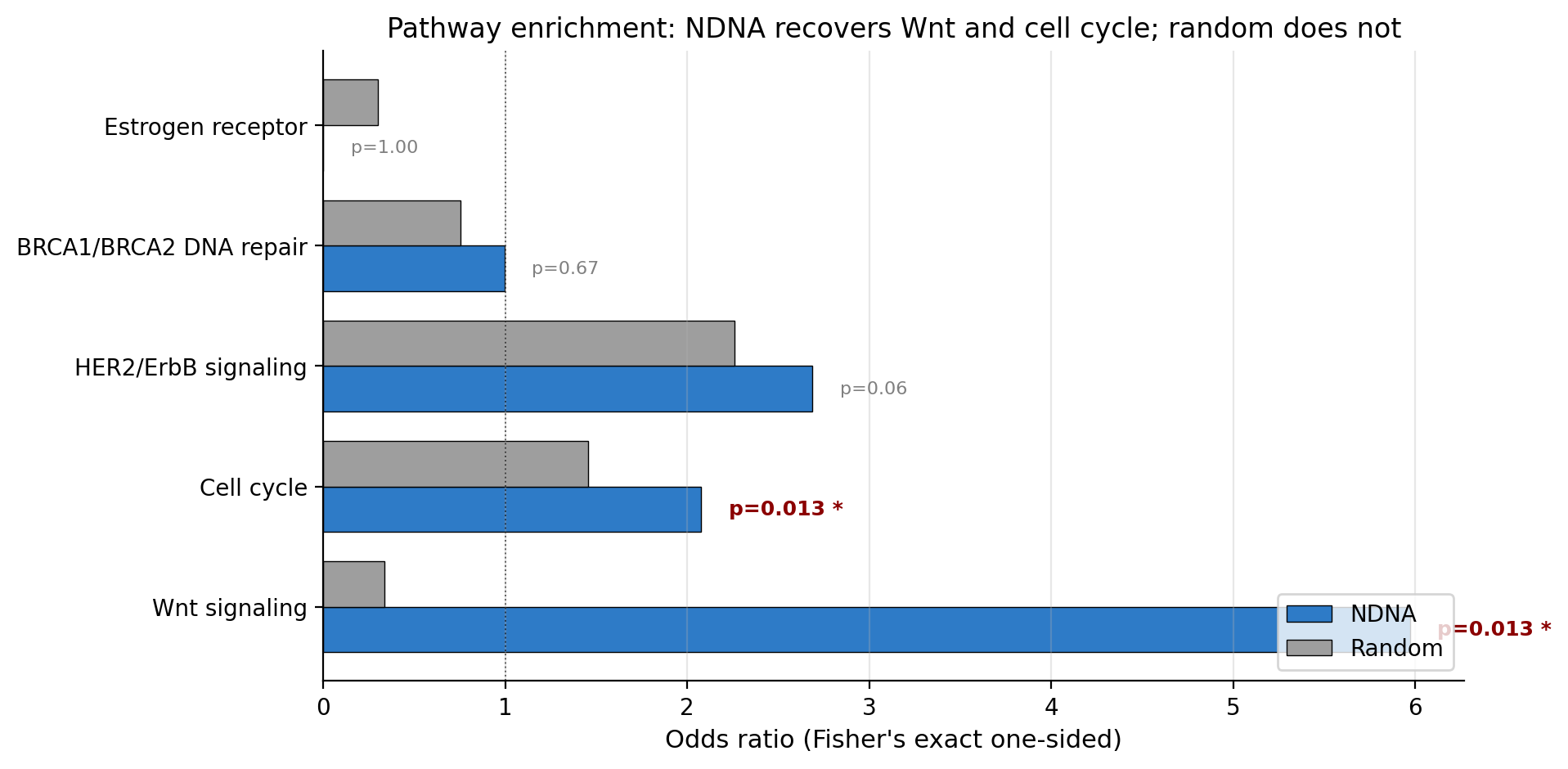
The genome's edge selection (blue) compared to a random control at the same density (gray). For Wnt and cell cycle, the difference is enormous. The genome was 18 times more likely than chance to keep a Wnt edge. Random selection would not have found these pathways.
Out of the 10 Wnt-pathway connections in our network, the genome kept 8 of them. Random selection at the same density would keep about 1 or 2. The probability of getting that kind of overlap by chance is 1.3% (Fisher's exact p = 0.013). Same story for cell cycle: the genome kept 25 out of 43 connections (p = 0.013); random would keep 14, which is not statistically significant.
So the genome rediscovered the most well-known cancer pathways without being told they exist. That is the headline.
There is a second finding in this same data that I find even more interesting.
What the Genome Refused to Keep
The estrogen receptor pathway is also famous in breast cancer biology, but for a different reason. Estrogen receptor (ER) status determines whether a tumor is "ER-positive" or "ER-negative". This affects which drugs work on the tumor. ER status is critical for treatment, but it does not distinguish tumor from healthy tissue. Healthy breast tissue also has estrogen receptors. The presence of ER signaling does not tell you "this is cancer". It tells you "this is breast tissue".
So if the genome was trained to distinguish tumor from healthy tissue, it should not care about ER signaling. The signal would be useless. Either you pick up ER edges in both, or in neither. They are not informative.
We had 11 estrogen receptor pathway connections in the network. The genome kept zero of them. It actively excluded the entire pathway.
Nobody told the genome this. We did not write any code that said "ignore ER". The genome figured out, from the data alone, that those connections did not help with the tumor-vs-healthy task. So it dropped them all.
That is the kind of behavior that suggests the genome is doing real biology, not just curve-fitting.
The Tumor Marker Surprise
Here is the most striking finding from inspecting individual connections.
After looking at the well-known pathways, we also looked at the highest-confidence connections the genome picked that were not in any of the seven cancer pathways we checked. These are the genome's "novel" picks. The list goes:
| Rank | Edge | What it is |
|---|---|---|
| 1 | LAMB3 - LAMC3 | Two laminin proteins; involved in tumor invasion |
| 2 | CACNA2D1 - CACNB4 | Calcium channel components |
| 3 | FZD9 - WNT3 | A Wnt receptor and a Wnt protein. Direct cancer relevance. |
| 4 | CEACAM5 - KLK3 | The genes for CEA and PSA, two clinically used tumor markers |
| 5 | CACNA2D1 - SCN3B | Calcium and sodium channel crosstalk |
| 6 | CEACAM5 - EPCAM | Two epithelial tumor markers |
| 7 | DSG3 - DSP | Desmosome components, often lost during metastasis |
Look at rank 4. CEACAM5 is the gene that makes a protein called CEA (carcinoembryonic antigen). KLK3 is the gene that makes a protein called PSA (prostate-specific antigen).
If you have ever had a blood test for cancer, those are probably the two markers your doctor measured. CEA is the standard blood marker for colon cancer, lung cancer, and several others. PSA is the standard blood marker for prostate cancer. These are not obscure proteins. They are some of the most clinically validated cancer biomarkers in existence.
The genome picked the interaction between them as one of its highest-confidence selections. Without being told that CEA or PSA are tumor markers. Without any supervision on tumor markers at all. It just inferred, from breast cancer expression data, that these two proteins talking to each other is meaningful.
Compare that to what a random control at the same density picked: the top connections were dominated by olfactory receptors and signaling generics. Random did not find a coherent oncology story.
This is the kind of result a biologist looks at and goes "huh, that is interesting". It does not mean we have proven anything wet-lab worthy. It means the genome is identifying connections that turn out to be biologically important, in a way that random selection does not.
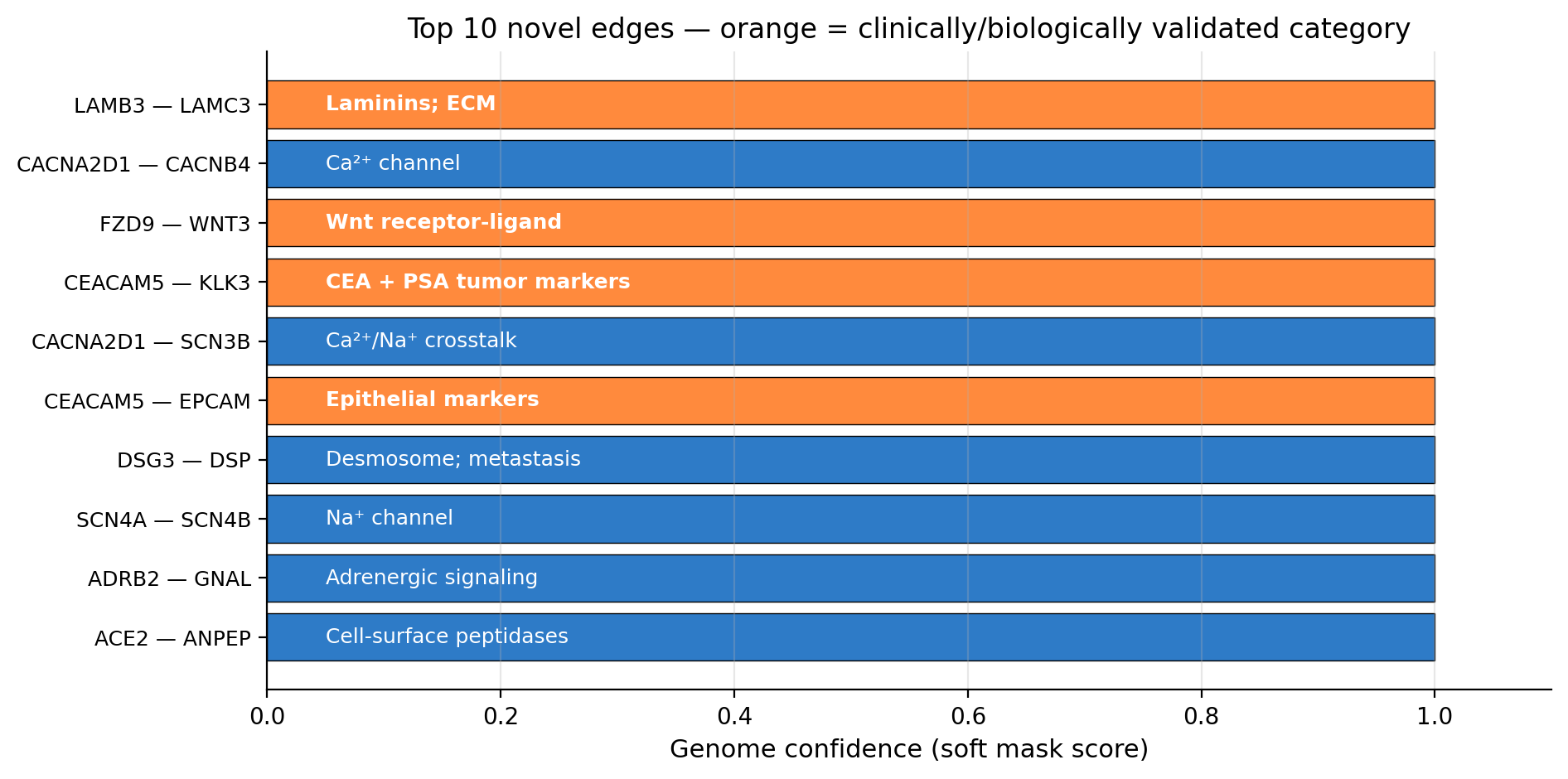
The genome's top 10 highest-confidence novel selections. Orange highlights the ones with strong clinical or pathway support: laminins involved in invasion, the Wnt receptor-ligand pair, two pairs of epithelial tumor markers including the famous CEA/PSA pair.
The Cross-Cancer Test
Here is the experiment I think is the most useful for actual cancer research.
Question: when the genome learns from breast cancer data, is it learning breast-cancer-specific tricks, or is it learning something general about cancer?
Test: take the genome trained on breast cancer, freeze it (do not let it learn anything new), and apply it to three completely different cancers: lung, colon, and prostate. Do the picked-out connections still distinguish tumor from healthy in those cancers?
We compared three setups for each cancer:
- Dense: use all the network connections (the standard approach)
- Frozen NDNA: use the connections the breast-cancer genome picked
- Random: pick the same number of connections at random
Run each of these five times with different random seeds, take the average and the spread. Here is what we got:
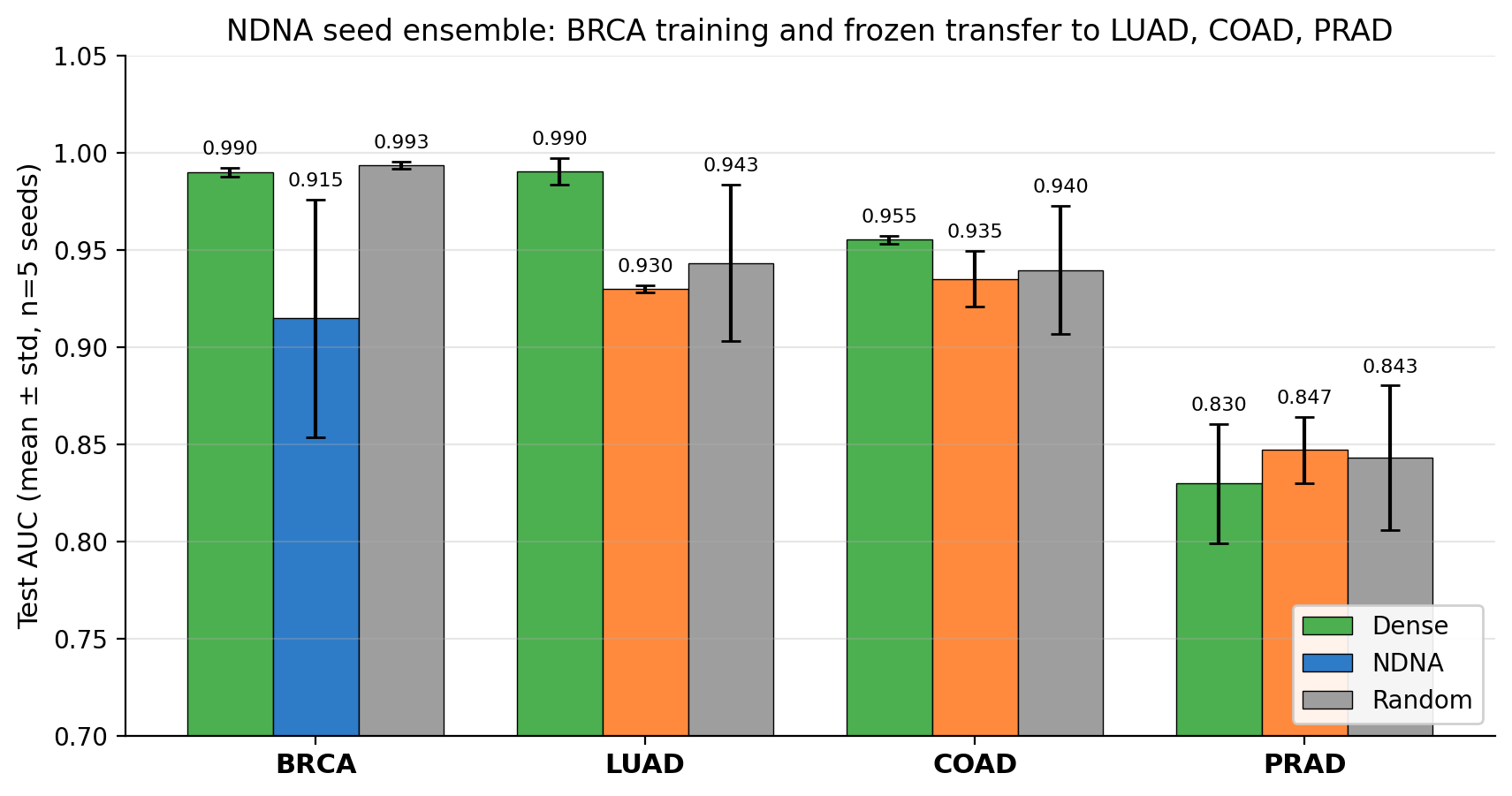
Each cancer (BRCA, LUAD, COAD, PRAD) tested in three conditions across 5 random seeds. Bars show mean test AUC, error bars show standard deviation across seeds. The frozen NDNA bars (orange) are competitive in mean and dramatically tighter in spread.
Two findings.
Finding 1. The frozen breast-cancer genome works on lung, colon, and prostate cancer. It gets results competitive with random selection at the same density, often slightly better. So the genome is not breast-specific. It learned something general about cancer.
Finding 2. This is the bigger one. The frozen genome is dramatically more reliable than random. Across five different random seeds, the frozen genome gives almost the same result every time. Random gives wildly different results depending on the seed.
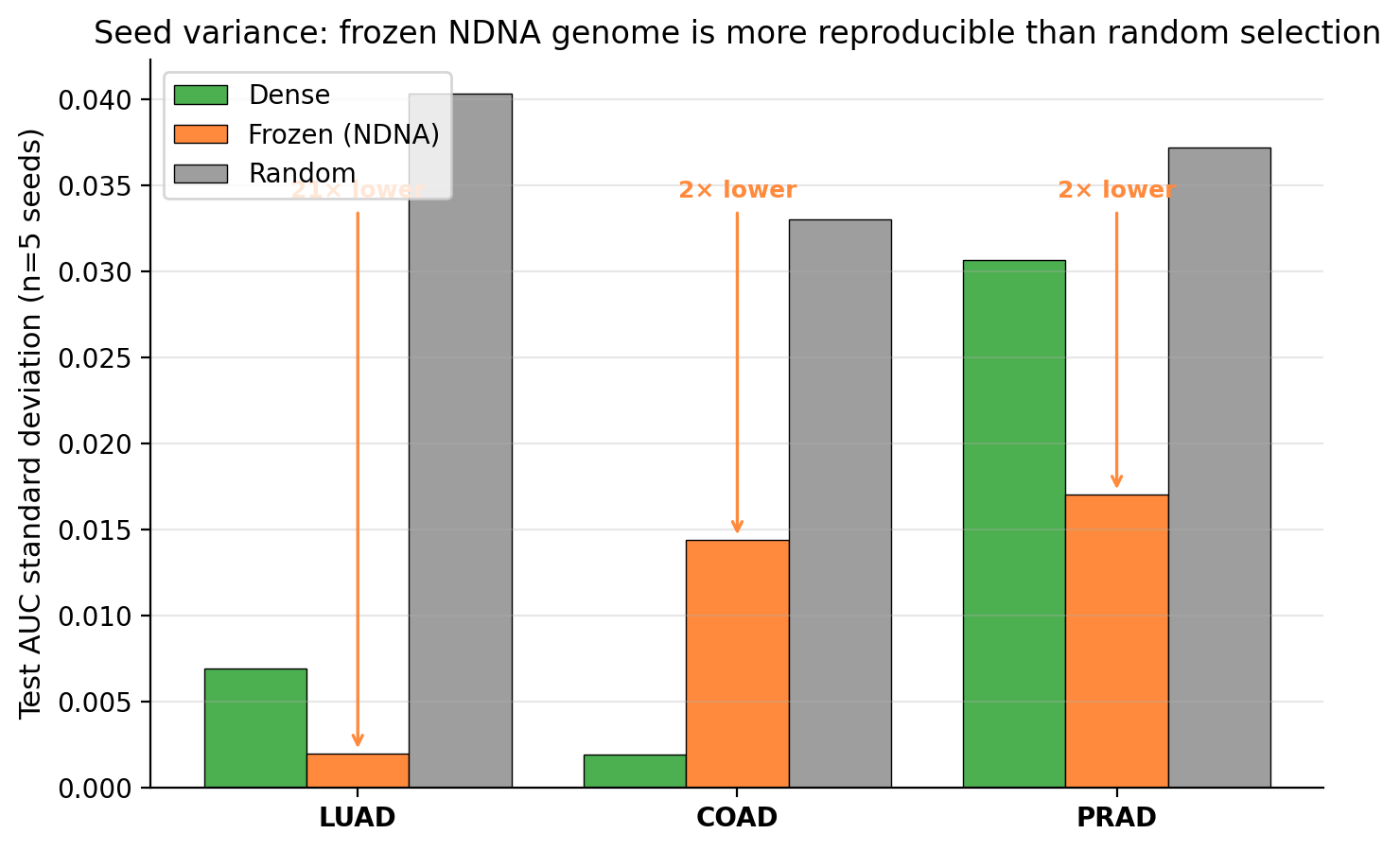
Standard deviation across seeds. Lower is better (means more reliable). Frozen NDNA wins in every cancer. On lung cancer, frozen is 21 times more reliable than random.
On lung cancer specifically: the frozen genome's spread across seeds is 0.002 in test AUC. Random's spread is 0.040. That is a 20 times reduction in variance. If you are a cancer researcher and you train a model, you want to know that the model gives the same answer when you run it again. Random sparse selection does not give that. The frozen NDNA genome does.
Why This Matters in Practice
Most computational cancer biology has a brutal data problem. Pancreatic cancer is one of the deadliest cancers in the world, but in the public TCGA database, there are only 4 healthy pancreatic tissue samples. You cannot train a reliable cancer-detection model with 4 healthy samples. You also cannot train it with 30. You probably need a few hundred. Most rare cancers have less than that. So those cancers do not get models, and they do not get the kinds of insights that come out of models. Patients suffer.
What if, instead of training from scratch on pancreatic cancer's tiny dataset, you took a genome trained on a data-rich cancer (like breast cancer, with 1,200+ patients) and just used its picked connections directly?
That is exactly what the cross-cancer transfer experiment shows is possible. A breast-cancer-trained genome gives you a stable, reproducible starting point for any other cancer you want to study. You are not curing pancreatic cancer with this. But you are giving a pancreatic cancer researcher a tool that the field currently lacks.
How It Connects to the Earlier Papers
In Paper 1, NDNA was a genome that grew the wiring of a neural network. The connections were inside an artificial brain.
In Paper 2, NDNA scaled to GPT-2. Same genome architecture. Same idea. Same story: a tiny program decides what gets connected to what.
In Paper 3, NDNA points at a completely different kind of network. Not artificial neurons. Real proteins. Real biology.
The genome architecture is essentially the same. 290 parameters. Eight types. A compatibility matrix. The same temperature schedule. The same idea: learn which connections to keep. Two small adaptations were needed (a different starting point for the genome, and a small change to the host network) and then it worked on biological networks.
This is the part that I find quietly significant about Paper 3. The genome abstraction is not a trick that only works for transformers. It travels. It works on networks that exist in nature.
If you want to see the math, the design choices, the failure modes, and the full hyperparameter sweep, the full paper is on the GitHub repo. The interactive visualization in Paper 2 shows what the GPT-2 version of the genome looks like during training.
What's Honestly Limited
A few things I want to be transparent about.
The genome is not magic on individual cancers. When we tried to train a fresh genome from scratch on lung, colon, or prostate cancer alone, the training was unstable. The breast cancer cohort is large enough (1,200 patients) for the genome to find a stable solution. Smaller cohorts are not. The frozen-transfer paradigm is what makes this useful.
No wet-lab validation. The CEACAM5–KLK3 finding is a computational hypothesis. Functional experiments would be needed to confirm whether disrupting that interaction matters for cancer. The right next step is partnering with a cancer biology lab.
Tumor vs healthy is a saturated task. Modern methods all do well at distinguishing cancerous from healthy tissue on TCGA data; the AUC ceiling on this benchmark is close to 1.0. The harder questions that actually move clinical practice (which subtype is this? will it metastasize? will this drug work?) are where computational biology has more room to contribute. We did not test those.
The genome's contribution here is not that it classifies cancer better than other methods. It does not. The contribution is that it identifies which protein interactions matter, in a way that recovers known biology and surfaces clinically validated markers, and that the same identification transfers across cancer types. That is interpretability and reliability, not raw accuracy.
Try It
git clone https://github.com/tejassudsfp/protein_study.git
cd protein_study
pip install -r requirements.txt
# Preprocess BRCA data (auto-downloads STRING + TCGA-BRCA)
python data/preprocess.py
# Train BRCA genome (dense, NDNA, random comparison)
python run.py --epochs 60
# Cross-cancer transfer test
python data/preprocess_cancer.py LUAD COAD PRAD
python run_transfer.py --cancers LUAD COAD PRAD --epochs 60
# Build the paper PDF (matches the look of Papers 1 and 2)
npm install
npm run build:pdf
Links
- Paper 3: Neural DNA on Protein Interaction Networks
- Code: GitHub
- DOI: 10.5281/zenodo.20026016
- Paper 1: Neural DNA: A Compact Genome for Growing Network Architecture
- Paper 2: Scaling Neural DNA to GPT-2 (with interactive viz)
Citation
@article{sudarshan2026ndnappi,
title = {Neural DNA on Protein Interaction Networks: A Compact Genome
Recovers Pan-Cancer Pathways and Transfers Across Cancer Types},
author = {Sudarshan, Tejas Parthasarathi},
year = {2026},
doi = {10.5281/zenodo.20026016},
url = {https://doi.org/10.5281/zenodo.20026016}
}